
Chimeric antigen receptor (CAR)-engineered T cells (CAR-T) have been remarkably successful in clinical treatment of relapsed or refractory haematological tumours such as B-cell acute lymphoblastic leukaemia (ALL), lymphomas and Multiple Myeloma1. Human induced pluripotent stem cells (iPSCs) are now poised to revolutionize the field of CAR-T immune cell therapy by offering a platform that can (a) be uniformly genetically engineered for allogeneic cell therapy and (b) offers a potentially unlimited source of T-cells for therapeutic applications2, that can help meet the increasing patient demand and clinical indications treatable by CAR-T3.
One of the key challenges in iPSC-based cell therapy is the controlled and efficient differentiation of iPSCs into specific cell lineages, particularly T-cells and their diverse sub-types (e.g., γδ T-cells, αβ CD4 or CD8 T-cells) (Figure 1, 4). Thus, the success of iPSC-based allogeneic cell therapies relies heavily on a comprehensive understanding of developmental and pluripotent stem cell biology and the ability to translate this to a robust cGMP manufacturing process.
This article explores the biological considerations involved in iPSC differentiation, with a specific focus on generating T-cells for cell therapy.
The power of iPSCs
iPSCs possess the unique ability to differentiate into any of the approximately 200 different cell types within the human body5. This capacity is harnessed for therapeutic purposes, aiming to supply specialized cells to regenerate damaged or dysfunctional tissues, or cells engineered with enhanced capacity to tackle disease (e.g., CAR-T cells).
However, achieving precise, efficient, and scaled differentiation of iPSCs into immune cells poses multifaceted biological challenges due to the intricate nature of the immune system.
Key signalling pathways for iPSC specification to T-cells
Differentiating iPSCs into T-cells cells requires the orchestration of specific signalling pathways, transcription factors, and epigenetic modifications that mimic the natural processes occurring during embryonic development.
Namely, FGF, TGF-beta family, and WNT signalling pathways, involved in mesodermal lineage formation, are key for initial differentiation of iPSC to hematopoietic stem and progenitor cells (HSPC, Figure 1)6. This process involves initial formation of hemogenic endothelium (HE) and subsequent definitive haematopoiesis via endothelial-to-hematopoietic transition (EHT)7. Later on, signal transduction pathways such as NOTCH as well as various interleukins regulate specification to lymphoid progenitors, proT-cells and mature T-cells.
Robust manufacturing processes that mimic natural developmental processes and microenvironment in the bone marrow and thymus are necessary to produce cells with the desired characteristics, emphasizing the importance of understanding the molecular and cellular events governing tissue-specific development. Natural developmental processes are mimicked in vitro by the addition of cytokines, growth factors, and small molecules, in a temporally and dose controlled manner. This helps guide iPSCs towards the desired immune cell fate. Understanding the intricate interplay of these factors is crucial for efficient and reproducible differentiation protocols.
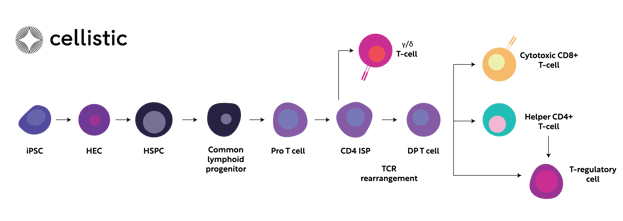
Figure 1. Simplified schematic representation of signalling pathways governing the step-wise specifications of iPSC towards diverse T-cell subtypes. iPSC; induced pluripotent stem cell, HEC; hemogenic endothelium, HSC; hematopoietic stem cell, IPS; immature single-positive, DP; double-positive, TCR; T-cell receptor
Lineage Commitment and Cell Fate Stability
Researchers are exploring strategies to enhance the stability of iPSC-derived T-cells, ensuring that they retain their intended functions over time, especially in the dynamic and complex immune microenvironment. An important consideration in iPSC differentiation is the establishment of a transcriptomic and epigenetic identity characteristic of the target cell type, which plays a pivotal role in maintaining the stable identity of differentiated cells.8
Epigenetic changes, including DNA methylation, histone modifications, and non-coding RNA expression, contribute to the stability of cell fate during lineage commitment. .Together with gene expression and phenotypic cell changes, epigenetic modifications and metabolism contribute to the long-term identity and functionality of differentiated T-cells. Therefore, preserving these precise cellular landscapes is crucial for achieving lineage commitment and cell fate stability, as well as avoiding undesired phenotypic drift of iPSC-derived cell types.
In vitro, this can be achieved by continuous monitoring and optimization of cell culture conditions that help lock the intended cell identity. Finally, robust in process and quality control measures need to be implemented to identify any cells maintaining an undifferentiated or progenitor cell state9 that might pose a safety risk for clinical use of allogeneic iPSC-based T-cell therapies.
Efficacy of iPSC-derived T-cell
Important advantages of iPSC-derived cytotoxic T-cells relate to the expansion that can be achieved in the manufacturing process from iPSC to T-cells, and their highly proliferative capacity compared to primary T-cells, which are easily exhausted and therefore a less than ideal starting point for allogeneic cell therapy.10
As well as having a high proliferation potential, antigen-specific cytotoxic T-cells derived from iPSCs reprogrammed from original antigen-specific (e.g., HIV-1) primary T-cells are powerful in killing virus-infected cell11. Similarly, EBV-specific iPSC-derived T-cells exert potent suppressive effects against EBV tumours.12
Challenges and Future Directions
Several iPSC allogeneic T-cell therapeutics are now in pre-clinical development and the first clinical trial utilizing iPSC-derived CAR-T cells is ongoing (NCT04629729).13
However significant challenges remain for therapeutic developers that want to enter the space.
- different cell lines necessitate clone selection14. Reports demonstrate a 25% frequency in isolating iPSC reprogrammed clones capable of reliable T-cell yield.15 Further improvements and standardization and automation of the process could improve this effect
- Continued exploration of high quality advanced animal-origin-free (AOF) defined media, feeder-free, and substrate-free methods to provide appropriate cell signalling are important to enabling clinical applications of iPSC-derived T-cell therapies.
- A deeper understanding of the dynamic interplay between signalling pathways and transcriptional networks is required to enhance the ability to control iPSC lineage commitment towards difficult to generate cell types (e.g., single-positive (SP) CD4+ T cells with full range of helper functions16) and ensure cell fate stability.
- The production of T cells has a multistep and lengthy nature, published protocols take approximately 7-10 weeks from iPSC establishment to hematopoietic T-cell specification and maturation to the fully functional effector cell-type (γ/δ, CD8, CD4) . This presents challenges in both process development & manufacturing.
- Scaling and closing available (academic) protocols to enable production of multiple, potentially thousands, of clinical doses is also a challenge. While controlled single-use stirred tank bioreactor systems are utilized for hematopoietic cells17 and T-cell expansion18, end to end scalable manufacturing processes have not been described.
Conclusion
The biological considerations in iPSC differentiation for cell therapy applications, particularly towards immune cells, are multifaceted. Therapeutic developers continue to unravel the complexities of guiding iPSCs through precise developmental pathways while ensuring the safety, stability, and functionality of the resulting cells. As advancements in iPSC technology progress, addressing these biological challenges will be pivotal in realizing the full potential of iPSC-based T-cell therapies, offering new avenues for treating haematological, but also solid tumour types, and immune-related disorders beyond oncology.
Sources:
1. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol (2023) 20(6):359–71. doi: 10.1038/s41571-023-00754-1
2. Netsrithong R, Garcia-Perez L and Themeli M (2024) Engineered T cells from induced pluripotent stem cells: from research towards clinical implementation. Front. Immunol. 14:1325209. doi: 10.3389/fimmu.2023.1325209
3. Biomedicines. 2021 Jan 9;9(1):59. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Ekaterina Zmievskaya 1, Aygul Valiullina 1, Irina Ganeeva 1, Alexey Petukhov 2, Albert Rizvanov 1, Emil Bulatov 1 3. doi: 10.3390/biomedicines9010059.
4. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel). 2016 Mar 15;8(3):36. doi: 10.3390/cancers8030036. PMID: 26999211; PMCID: PMC4810120.
5. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007 Nov 30;131(5):861-72. doi: 10.1016/j.cell.2007.11.019. PMID: 18035408.
6. Nostro MC, Cheng X, Keller GM, Gadue P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell (2008) 2(1):60–71. doi: 10.1016/j.stem.2007.10.011
7. Ottersbach K. Endothelial-to-haematopoietic transition: an update on the process of making blood. Biochem Soc T. (2019) 47:591–601. doi: 10.1042/BST20180320
8. Poetsch MS, Strano A, Guan K. Human Induced Pluripotent Stem Cells: From Cell Origin, Genomic Stability, and Epigenetic Memory to Translational Medicine. Stem Cells. 2022 Jun 22;40(6):546-555. doi: 10.1093/stmcls/sxac020. PMID: 35291013; PMCID: PMC9216482.
9. Watanabe T, Yasuda S, Kusakawa S, Kuroda T, Futamura M, Ogawa M, et al. Multisite studies for validation and improvement of a highly efficient culture assay for detection of undifferentiated human pluripotent stem cells intermingled in cell therapy products. Cytotherapy. 2021;23: 176–183. doi: 10.1016/j.jcyt.2020.07.009
10. Nishimura T., Kaneko S., Kawana-Tachikawa A., et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation.Cell Stem Cell. 2013; 12: 114-126
11. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
12. Ando M, Nishimura T, Yamazaki S, Yamaguchi T, Kawana-Tachikawa A, Hayama T, Nakauchi Y, Ando J, Ota Y, Takahashi S, Nishimura K, Ohtaka M, Nakanishi M, Miles JJ, Burrows SR, Brenner MK, Nakauchi H. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Reports. 2015 Oct 13;5(4):597-608. doi: 10.1016/j.stemcr.2015.07.011. Epub 2015 Aug 28. PMID: 26321144; PMCID: PMC4624898.
13. Chang CW, van der Stegen S, Mili M, Clarke R, Lai YS, Witty A, et al. FT819: translation of off-the-shelf TCR-less trac-1XX CAR-T cells in support of first-of-kind phase I clinical trial. Blood (2019) 134. doi: 10.1182/blood-2019-130584
14. Cahan P., Daley G.Q. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat. Rev. Mol. Cell Biol. 2013;14:357–368
15. Nagano S, Maeda T, Ichise H, Kashima S, Ohtaka M, Nakanishi M, Kitawaki T, Kadowaki N, Takaori-Kondo A, Masuda K, Kawamoto H. High Frequency Production of T Cell-Derived iPSC Clones Capable of Generating Potent Cytotoxic T Cells. Mol Ther Methods Clin Dev. 2019 Dec 24;16:126-135. doi: 10.1016/j.omtm.2019.12.006. PMID: 31970197; PMCID: PMC6965501.
16. Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, et al. Organoid-Induced Differentiation of Conventional T Cells From Human Pluripotent Stem Cells. Cell Stem Cell (2019) 24(3):376–89.e8. doi: 10.1016/j.stem.2018.12.011
17. Mohseni, S., Khoshfetrat, A.B., Rahbarghazi, R. et al. Influence of shear force on ex vivo expansion of hematopoietic model cells in a stirred tank bioreactor. J Biol Eng 17, 38 (2023). https://doi.org/10.1186/s13036-023-00358-4
18. Gatla, H. et al. Enabling Allogeneic T Cell-Based Therapies: Scalable Stirred-Tank Bioreactor Mediated Manufacturing. Front. Méd. Technol. 4, 850565 (2022).